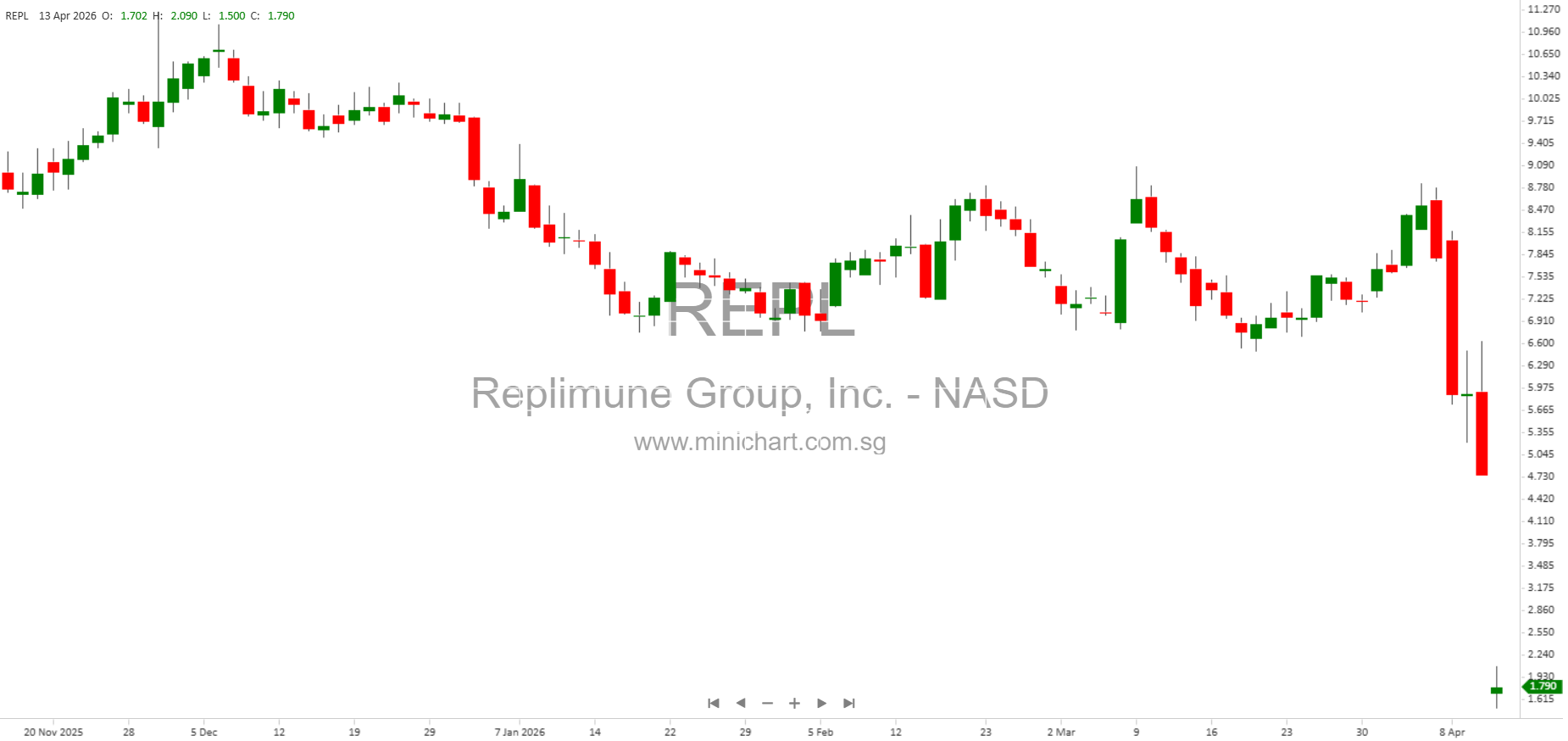
Replimune Receives FDA Complete Response Letter for RP1 Biologics License Application in Advanced Melanoma
Key Highlights and Details for Investors
Replimune Group, Inc. (NASDAQ: REPL), a clinical stage biotechnology company specializing in novel oncolytic immunotherapies, has announced a significant regulatory setback. On April 10, 2026, the company received a Complete Response Letter (CRL) from the U.S. Food and Drug Administration (FDA) regarding its Biologics License Application (BLA) for RP1 (vusolimogene oderparepvec) in combination with nivolumab for the treatment of advanced melanoma.
Key Points from the Report
- The FDA did not approve the BLA for RP1 in advanced melanoma. The CRL signals that the FDA does not believe the current data package is sufficient for approval, despite RP1’s prior breakthrough therapy designation and promising trial results.
- IGNYTE Trial Data: The pivotal IGNYTE trial demonstrated a 34% response rate in patients with advanced melanoma who had progressed on prior anti-PD-1 therapy, with a median duration of response of 24.8 months and a favorable safety profile.
- Company Disagrees with FDA’s Assessment: Replimune expressed disappointment, emphasizing the urgent unmet need for new treatments in advanced melanoma (which causes approximately 8,500 deaths per year in the U.S.). The company criticized the FDA’s lack of regulatory flexibility and communication, emphasizing that melanoma specialists and patients advocated for the therapy.
- Impact on Operations: Due to the lack of timely accelerated approval, Replimune announced that further development of RP1 will not be viable. The company will eliminate jobs and substantially scale back U.S.-based manufacturing operations. This restructuring is a direct result of the regulatory decision, not a failure of the therapy itself.
Details of Regulatory Interactions and Disputes
- Change in FDA Review Team: The company learned that a different FDA review team was appointed for the resubmission, replacing the prior team. According to Replimune, a senior member of the previous team stated that the clinical team found the evidence sufficient, but FDA leadership did not agree.
- Inconsistent FDA Communication: The company claims the FDA contradicted its prior positions, including acceptance of the single-arm trial design and the sufficiency of the data set, which had led to breakthrough therapy designation and priority review.
- Trial Design and Data: Replimune followed FDA guidance in trial design and data analysis, including using RECIST 1.1 criteria for tumor assessment. The company provided additional analyses as requested, including data on progression-free survival and response rates between injected and non-injected lesions.
- Accelerated Approval Pathway: The FDA previously indicated that a sufficiently compelling single-arm trial could be considered for accelerated approval, and the company had launched a global Phase 3 trial (IGNYTE-3) as a confirmatory study.
Potential Impact on Shareholders
- Material Adverse Impact: The FDA’s CRL is a major regulatory setback, likely to have a significant negative impact on Replimune’s share price due to the loss of near-term commercial opportunity for RP1 in advanced melanoma and the decision to scale back operations.
- Job Cuts and Manufacturing Reduction: The company will be eliminating jobs and reducing U.S. manufacturing, which indicates a substantial downsizing and cost-cutting mode, raising questions about the future of other programs in the pipeline.
- Regulatory Risk and Uncertainty: The situation highlights high regulatory risk and uncertainty around future interactions with the FDA, especially for therapies in rapidly evolving and high-need cancer indications.
- Loss of Breakthrough Therapy Momentum: The failure to secure accelerated approval, even with breakthrough therapy designation and positive clinical data, may undermine investor confidence in the company’s ability to bring new therapies to market.
Background on Melanoma and RP1
Melanoma is the fifth most common cancer in the U.S., with about 112,000 new cases and nearly 8,500 deaths expected in 2026. Standard care includes immune checkpoint inhibitors, but about half of patients do not respond or relapse. RP1 is an engineered herpes simplex virus therapy designed to kill tumor cells and stimulate a systemic immune response, intended to work synergistically with other cancer treatments.
Forward-Looking Risks
Replimune’s press release includes forward-looking statements about regulatory interactions, the future of the RP1 program, and operational changes. The company cautions that actual outcomes may differ due to risks such as regulatory hurdles, operational challenges, competition, and macroeconomic factors.
Contact Information
- Investor Inquiries: Chris Brinzey, ICR Healthcare, 339.970.2843, [email protected]
- Media Inquiries: Arleen Goldenberg, Replimune, 917.548.1582, [email protected]
Disclaimer: This article is for informational purposes only and does not constitute investment advice. Investors should conduct their own research and consult with their financial advisor before making investment decisions. The situation described involves significant regulatory and operational uncertainty, and actual outcomes may differ from those projected. The author and publisher are not responsible for investment outcomes or actions taken based on this article.